Building models from adsorption complexes
To create a model of some adsorption system that can be loaded by the susmost.load_lattice_task() function one needs structures of adsoption complexes (AC) and of the clean surface slab specified in Extended XYZ format. Several ACs are provided with SuSMoST in acs directory and can be downloaded from Downloads or from Adsorption Complexes. If you want to simulate some system with other ACs, then you can make them on you own as described in Creating sketches of a surface slab and adsorption complexes.
Making pairwise samples
SuSMoST’s adsorption models are essentially directories populated by files that describe various model parameters: lattice site states, possible interactions, interaction energies, samples of pairwise configurations and single adsorption complexes. Although it is possible in principle to create a model directory manually, this is a very laborous and error prone task.
Much simpler way is to use a special command line utility make-ac-samples. This utility is a part of SuSMoST and is located in utils/ subdirectory of SuSMoST distribution archive.
To use this utility you need an acitvated virtual environment with installed SuSMoST Python packages. See usage of source command in Running simulations for details. The typical call of the make-ac-samples utility is the following
./utils/make-ac-samples -X 5 -Y 5 -d CO-Pt111 acs/empty-Pt111.xyz acs/CO-atop-Pt111.xyz
Here -X 5 and -Y 5 parameters set the size of generated pairwise samples along X and Y axises correspondingly. The -d CO-Pt111 parameter sets the destination model directory where generated samples should be saved. The list of arguments should end with the list of files with adsorption complexes: acs/empty-Pt111.xyz acs/CO-atop-Pt111.xyz. The first file should be the empty slab sketch. Subsequent files are adsoption complexes to be considered in the model.
The make-ac-samples utility script (as well as the susmost.make_ac_samples() function that it wraps) creates the following files in the destination model directory:
sample_empty_unit_cell.xyz- xyz-file with surface unit cell;
sample_ac_{AC-name}_{index}.xyz- xyz-files with single adsorption complexes, here {AC-name} is a name of adsorption complex, {index} is an index that enumerate slabs of different sizes;
sample_{index}.xyz- xyz-files with pairs of adsorption complexes;
interactions- matrices of pairwise interactions that essentially describes symmetry of the model hamiltonian derived from geometrical symmetries of empty surface slab and adsorption complexes;
adsorbate_state_{index}.xyz- files for visualization of lattice site states.
Computing and specification of lateral interaction energies
When surface samples are created, their energies should be specified in the file named energies. This file should be placed in the same folder along with surface samples. Generally these energies can be computed in any way. The format of the energies file is straightforward and is formally described in reference of the fucntion: susmost.load_lattice_task(). Here is an example from the TPA-Fe/Cu(100) model (Example of Model of Fe-Terephthalate Ordering on Cu(100) : Monte Carlo method):
sample_030.xyz = 4.0 sample_034.xyz = -4.5 sample_044.xyz = -3 sample_083.xyz = inf sample_084.xyz = 12.0
Here inf means that the corresponding configuration is unstable and therefore forbidden in this model. Skipped samples correspond to configurations with negligible lateral interactions. Energies can also be specified for samples with single ACs and with empty slab. In that case lateral interaction’s energies will be derived by substracting of slab’s and AC’s contributions from total energy of pairwise samples.
Energies can be computed with script utils/calc-energies. Typical usage is the following:
./utils/calc-energies -d ./models/NH3-V3C2 --hsph-r N=2.0
This command computes lateral interactions between adsorption complexes of the model in NH3-V3C2 directory using hard-sphere calculator. Argument --hsph-r N=2.0 means that hard-sphere radius of nitrogen atoms should be 2.0 angstrom. All other elements are ignored here. These command creates the energies file with only one row:
sample_001.xyz = inf
The sample in file sample_001.xyz contains the following configuration:
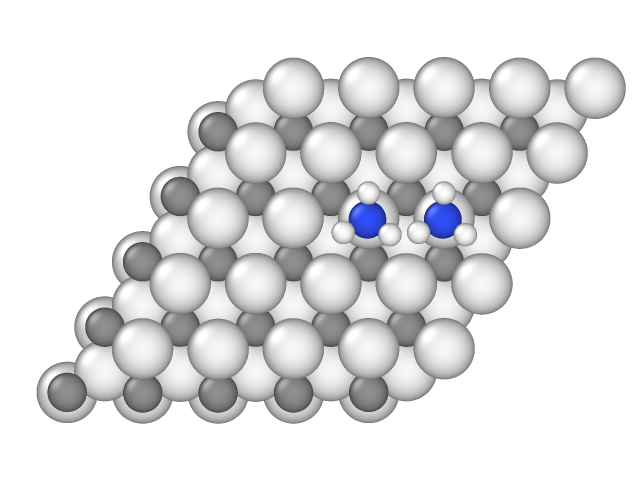
Here distance between nitrogen atoms is 2.95 angstrom, therefore their hard spheres with 2.0 angstroms radius intersect, and this configuration is forbidden. In other pair samples of the NH3-V3C2``model the distance between nitrogen atoms is larger than 4.0 angstrom, therefore there are no lateral interactions, and these configurations are not included in ``energies file.
To help you with energies calculation there are exemplary python scripts in SuSMoST that compute energies of surface samples with ASE calculators - either QuantumEspresso or GPAW, and write them into the energies file in proper format.