Tutorial - Transfer-matrix simulation Langmuir adsorption model with interaction
In this tutorial we provide an example of applying the transfer-matrix method to the Langmuir adsorption model with interaction between nearest neighbors. SUrface Science MOdeling and Simulation Toolkit (SuSMoST) is a set of computer programs and libraries that are used for modeling. As a result of the program, we get adsorption isotherm \(\theta(\mu)\).
Please have a look at the following script here and read below what the script does.
Libraries
In this block we include libraries. Here numpy is standard library. susmost is a general library that contains modules required for setting structure of model and applying the Transfer-matrix method.
import numpy as np
from susmost import make_single_point_zmatrix, make_square_plane_cell, \
normal_lattice_task, transferm, nearest_int, SiteStateType, make_tensor, solve_TM
Constants
Here we define the constants.
INF_E = 1E6
k_B = 8.3144598E-3
a = 1.0
r_cut_off= 8.0 + .0001
In the table below, we show the constants and their descriptions.
Constant |
Description |
|---|---|
INF_E |
Prohibitive energy level |
k_B |
Gas constant, kJ/(mol*K) |
a |
Lattice constant |
r_cut_off |
Constant required to calculate the transfer-matrix |
Note
If k_B is measured in kJ/K, then k_B is the Boltzmann constant .
Parameters
Here we define the parameters.
N = 8
T = 200.0
mol_int = 2.5
In the table below, we show the parameters and their descriptions.
Parameter |
Description |
|---|---|
N |
Width of the semi-infinite system (infinite in one direction and finite in perpendicular one) |
T |
Temperature, K |
mol_int |
Interaction energy, kJ/mol |
Here we set the lattice geometry that models the solid surface.
cell,atoms = make_square_plane_cell(a)
interaction = lambda cc1,cc2: nearest_int(cc1,cc2,mol_int)
In this models we use a square lattice with lattice parameter a (make_square_plane_cell(a) and etc.). The lattice constant a specifies the location of the nodes.
interaction define an interaction between neighbor sites.
Initial data
In this block we initialize input data.
Here mus - array of chemical potential differences in the gas and adsorption layer \(\mu_g - \mu_a \approx-RTlnp\), covs - array of coverages, \(\beta=1/(k_B T)\).
beta = 1./(k_B*T)
mus = []
covs = []
Calculation of coverages
Set interval in what we change the pressure in the gas phase. mono_state and empty_state specifies the shape of the nodes (make_single_point_zmatrix()), sets for each node the energy of adsorption ( monomer - mu, empty - 0), assigns the mark to all the possible surface states (monomer - atom N, empty - H), sets the properties of each node (monomer - coverage=1., empty - coverage=0.).
The lattice model description is stored in lt as objects of the class LatticeTask.
Set W as a tensor of interaction that can be used to solve eigenvalue problem [1] (read more: interface round a face [2] ), avg_cov - the average cover corresponding to each ring and tm_sol as solution of transfer-matrix.
In result we’ll get an array of the calculated coverages by transfer-matrix method covs and the chemical potential mus.
for mu in np.arange(-30., 20.+0.0001, 2.0 ):
mono_state = SiteStateType('mono', make_single_point_zmatrix(), mu, ['N'], coverage=1.)
empty_state = SiteStateType('Empty',make_single_point_zmatrix(), 0.0, ['H'], coverage=0.)
lt = normal_lattice_task([mono_state, empty_state], cell, atoms, interaction, r_cut_off, INF_E)
W = make_tensor(lt, beta)
avg_cov = transferm.average_props(lt.states, N, 'coverage', beta)
tm_sol = transferm.solve_TM(W, N, symmetric=True)
cov = sum(tm_sol.probs*np.array(avg_cov))
covs.append(cov)
mus.append(mu)
print ("mu ==", mu, "theta == ", cov)
Examples
Let’s look at some examples of using the application by varying input data, such as:
energy of interaction
mol_int;step of adsorption energy
mu;temperature
T.
Note
We considered examples for the width of the lattice N=6. In case of necessary it can be changed too.
Example 1.
Assume we have next initial data:
Data |
|---|
T = 100 |
mu = (-50,50,1) |
here step_mu=1.
Let change the value of interaction energy mol_int. As result we get the following adsorption isotherms (fig. 1)
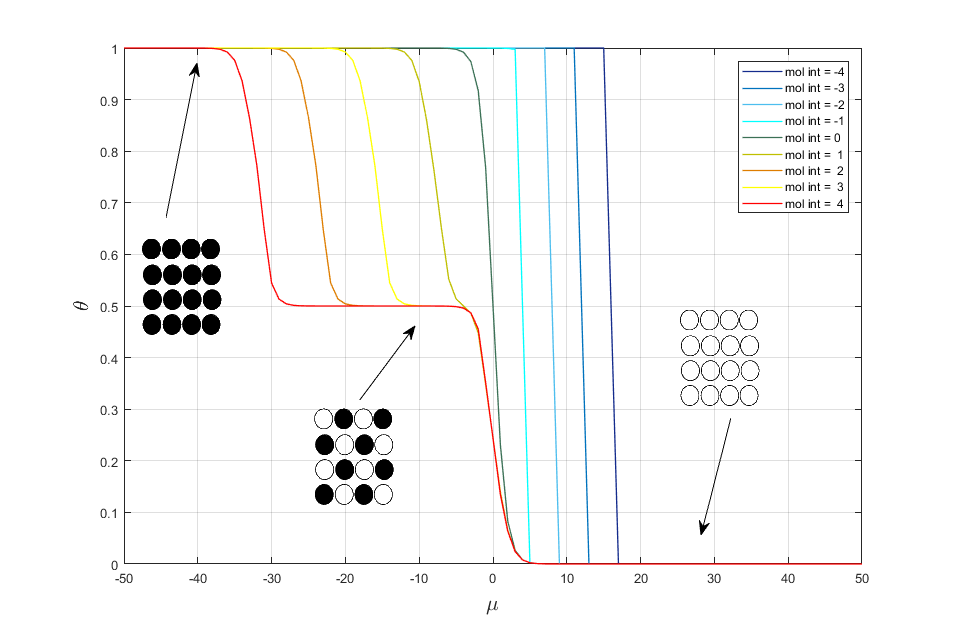
Fig. 1. Adsorption isotherms for different interaction energy.
It is seen from fig. 1 that in case of attraction we get the two-dimensional condensation and in the case of repulsion we get two phase transitions with forming of a chess and dense phases. If mol_int = 0, then we get classical Langmuir model.
Example 2.
Assume we have next initial data:
Data |
|---|
T = 100 |
mol_int = 4 |
mu = (-10,10) |
Let change the step of adsorption energy mu. As result we get the following adsorption isotherms (Fig. 2)
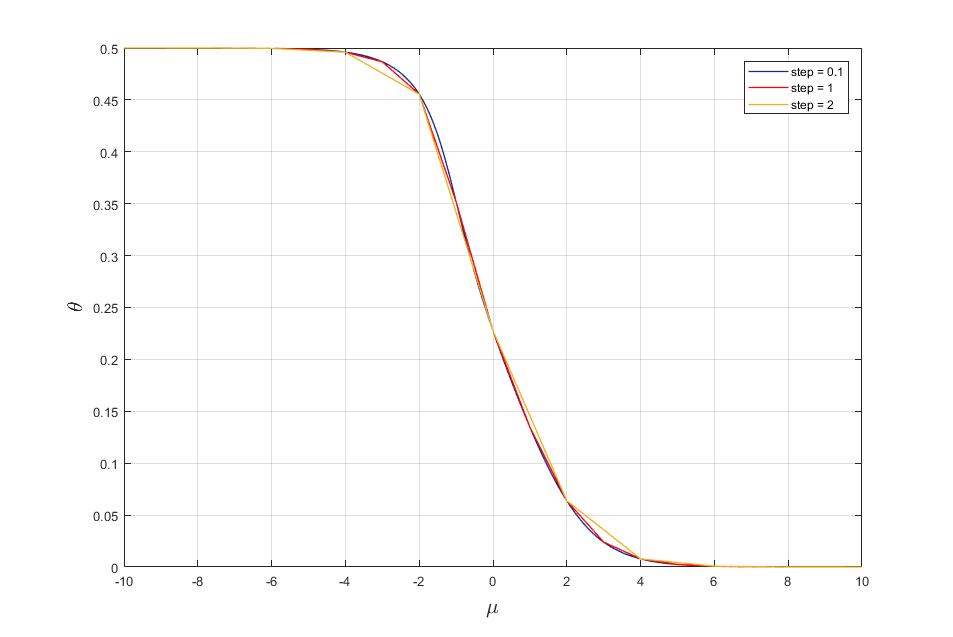
Fig. 2. Adsorption isotherms for mu = (-10,10) with different step.
It is seen from fig. 2 that by changing the step of adsorption energy mu we can obtain adsorption isotherms with a certain accuracy.
Example 3.
Assume we have next initial data:
Data |
|---|
mu = (-50,50,1) |
mol_int = 4 |
Let change values of temperature T. As result we get the following adsorption isotherms (Fig. 3)
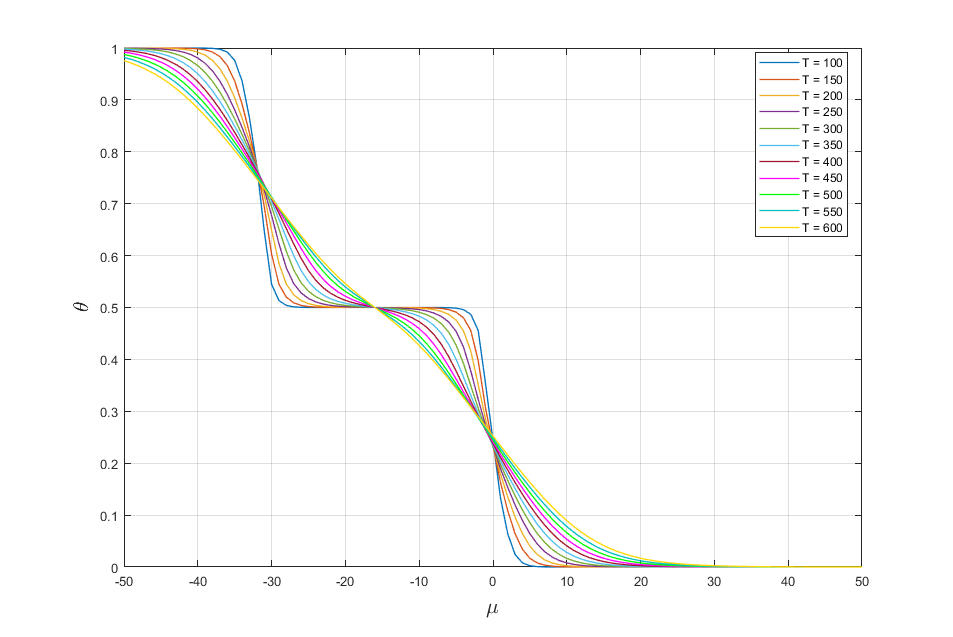
Fig. 3. Adsorption isotherms for different values of temperature.
It is seen from fig. 3 that with increasing temperature the region of existence of the phases are reduced to their disappearance.